Diffusion-based structure prediction can be guided by backpropagating to the conditioning embeddings rather than the atomic coordinates directly, and such embeddings can be re-refined in subsequent iterations
Diffusion-based biomolecular structure prediction, which is used in latest-generation methods like AlphaFold3[1] and BioEmu[2], can be guided or steered into specific conformations by backpropagating to the conditioning representations rather than the atomic coordinates being diffused [3][4]. This was recently shown by two methods, EmbedOpt and IT-Optimization.
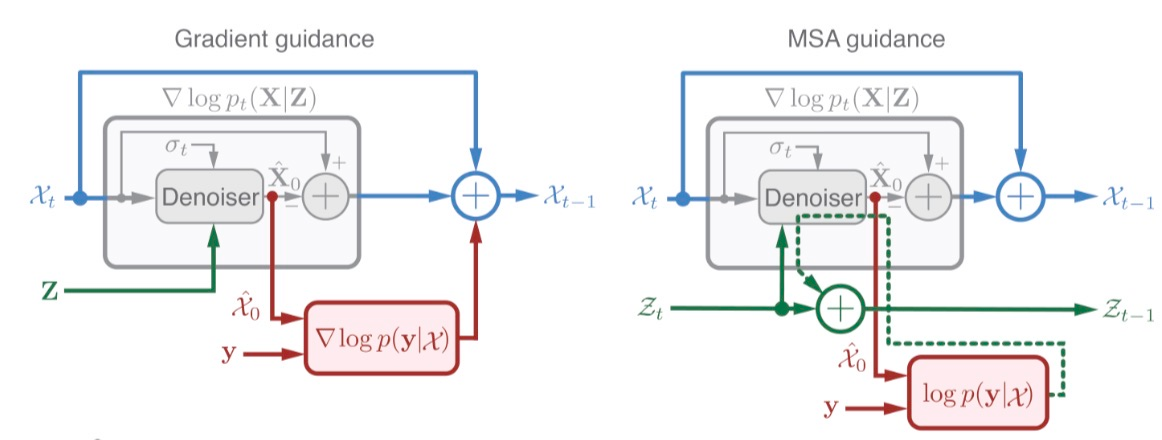
There are two arguments for doing this over standard Diffusion Posterior Sampling[5] (DPS), which operates directly on the elements being subject to diffusion (here, atomic coordinates). First, DPS tends to distort the outputs and send them off the manifold of realistic conformations. This is consistent with my own experience with cryoBoltz[6], which uses DPS; often, for example, chainbreaks are introduced to occupy spurious density clouds.
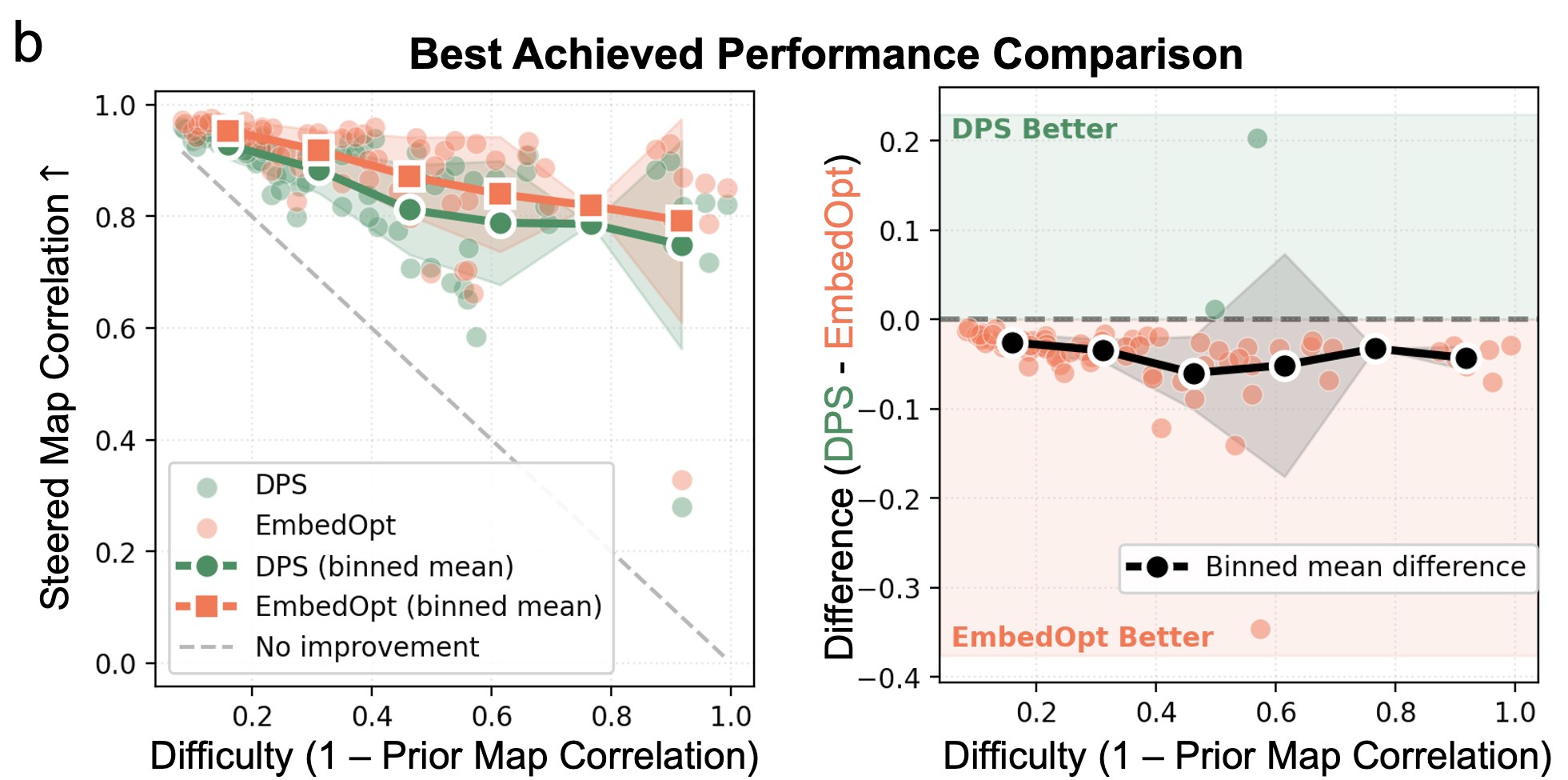
Second, the conditioning embeddings can be re-used and re-refined in subsequent runs, leading to even greater improvements. The figure below shows how a single run of Protenix (labeled AF3) can match or even exceed guidance of conditioning representations, but is fundamentally limited to a single diffusion roll-out; subsequent inference must start from scratch.
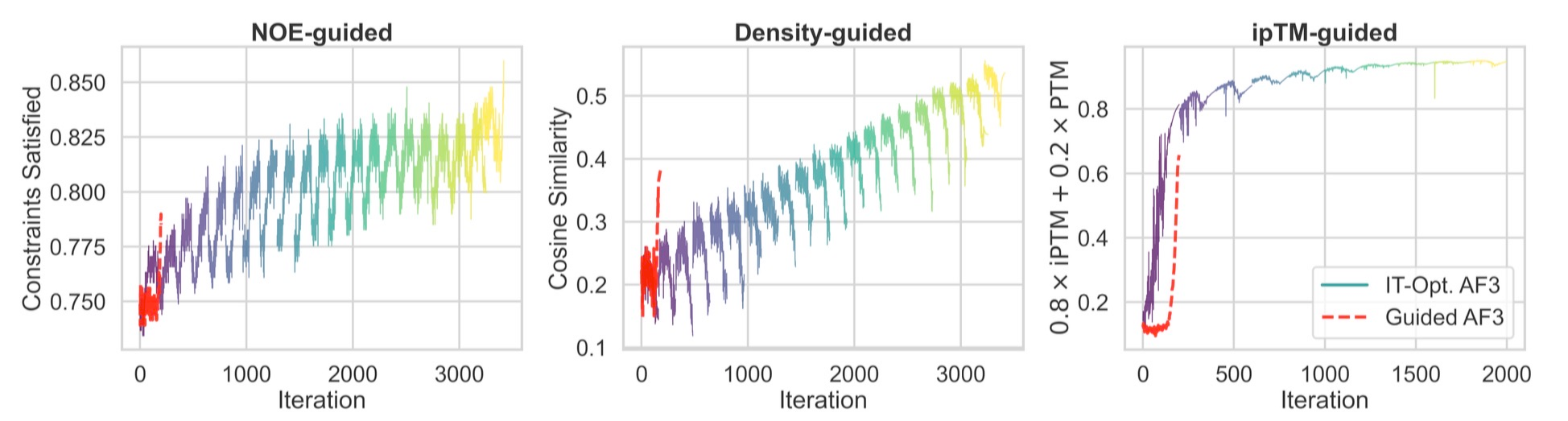
As the recent report by Nvidia discusses[7], such guidance approaches begin to blend together elements of hallucination-based and diffusion-based protein modeling.
References:
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A. J., Bambrick, J., Bodenstein, S. W., Evans, D. A., Hung, C.-C., O’Neill, M., Reiman, D., Tunyasuvunakool, K., Wu, Z., Žemgulytė, A., Arvaniti, E., … Jumper, J. M. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630(8016), 493–500. https://doi.org/10.1038/s41586-024-07487-w ↩︎
Lewis, S., Hempel, T., Jiménez-Luna, J., Gastegger, M., Xie, Y., Foong, A. Y. K., Satorras, V. G., Abdin, O., Veeling, B. S., Zaporozhets, I., Chen, Y., Yang, S., Foster, A. E., Schneuing, A., Nigam, J., Barbero, F., Stimper, V., Campbell, A., Yim, J., … Noé, F. (2025). Scalable emulation of protein equilibrium ensembles with generative deep learning. Science, 389(6761). https://doi.org/10.1126/science.adv9817 ↩︎
Li, M., Han, J., Cossio, P., & Wu, L. (2026). Robust Inference-Time Steering of Protein Diffusion Models via Embedding Optimization (Version 1). arXiv. https://doi.org/10.48550/ARXIV.2602.05285 ↩︎
Maddipatla, A., Rzayev, A., Pegoraro, M., Pacesa, M., Schanda, P., Marx, A., Vedula, S., & Bronstein, A. M. (2026). Inference-time optimization for experiment-grounded protein ensemble generation (Version 2). arXiv. https://doi.org/10.48550/ARXIV.2602.24007 ↩︎
Chung, H., Kim, J., Mccann, M. T., Klasky, M. L., & Ye, J. C. (2022). Diffusion Posterior Sampling for General Noisy Inverse Problems (Version 4). arXiv. https://doi.org/10.48550/ARXIV.2209.14687 ↩︎
Raghu, R., Levy, A., Wetzstein, G., & Zhong, E. D. (2025). Multiscale guidance of protein structure prediction with heterogeneous cryo-EM data (Version 2). arXiv. https://doi.org/10.48550/ARXIV.2506.04490 ↩︎
Didi, K., Zhang, Z., Zhou, G., Reidenbach, D., Cao, Z., Cha, S., Geffner, T., Dallago, C., Tang, J., Bronstein, M. M., Steinegger, M., Kucukbenli, E., Vahdat, A., & Kreis, K. (2026). Scaling atomistic protein binder design with generative pretraining and test-time compute. The Fourteenth International Conference on Learning Representations. https://openreview.net/forum?id=qmCpJtFZra ↩︎